|
|
|
|
 |
|
5 ans après un AVC bien traité, quels sont les risques de récidive ? |
|
|
| |
|
| |

5 ans après un AVC bien traité, quels sont les risques de récidive ?
COMMUNIQUÉ | 23 MAI 2018 - 16H18 | PAR INSERM (SALLE DE PRESSE)
PHYSIOPATHOLOGIE, MÉTABOLISME, NUTRITION
L’équipe du Pr Amarenco, chef du service de neurologie à l’hôpital Bichat Claude-Bernard, AP-HP, professeur à l’université Paris Diderot – Inserm, a étudié le risque de récidive d’un AVC pour des patients ayant bénéficié d’une prise en charge dans les 24 heures suivant la survenue d’un accident ischémique transitoire. Les chercheurs démontrent que de 1 à 5 ans le risque reste constant. Ces résultats suggèrent que la prévention de ces événements ne doit pas se concentrer que durant les premiers mois suivant l’AVC, mais doit être prolongée au moins 5 ans.
Ils sont publiés dans le New England Journal of Medicine le 16 mai.
Aujourd’hui, environ un accident cardio-vasculaire sur quatre est précédé d’un accident ischémique transitoire. Il peut par exemple se manifester par une paralysie d’un membre, une perte de la parole et/ou de la vue ou encore des troubles de l’équilibre. Après un AIT ou un infarctus cérébral mineur (ne donnant pas de handicap immédiat), le risque à long-terme de survenue d’un autre AVC, d’un infarctus du myocarde ou d’un décès d’origine vasculaire n’est pas connu.
Après avoir rapporté lors d’une première étude parue en 2016 dans le New England Journal of Medicine, le risque de survenue d’un accident cardio-vasculaire à un an, l’équipe s’est attachée à en mesurer le risque à 5 ans. Ces travaux ont été réalisés dans le cadre du projet international TIAregistry.org.
L’étude a été menée auprès de 3 847 patients issus de 21 pays (en Europe, Asie, Japon, Amérique Latine) entre 2009 et 2011, victimes d’un AIT ou d’un accident cérébral mineur dans les 24 heures pour 80% d’entre eux et pour les autres dans les 7 jours. L’objectif était notamment d’évaluer l’état de santé des patients, pris en charge dans une structure spécialisée, et le risque d’AVC cinq ans après la survenue de l’AIT ou d’un accident cérébral mineur. Parmi les 61 centres initiaux qui ont permis la première publication des données à 1 an, 42 ont participé au suivi des patients jusqu’à la 5ème année.
Parmi les 3 847 patients suivis 5 ans, 469 ont eu un infarctus cérébral, un infarctus du myocarde ou sont morts de problème vasculaire, soit un risque à 5 ans de 12,9%.
La moitié de ces événements est survenu au cours de la première année de suivi, la moitié est survenue entre la deuxième et la cinquième année, ce qui montre que la prévention de ces événements ne doit pas se concentrer que durant les premiers mois suivant l’AVC, mais doit être prolongée au moins 5 ans.
La survenue de ces événements reste constante au fil du temps, c’est-à-dire que le risque n’a pas tendance à s’atténuer.
À 5 ans, le risque de récidive d’AVC était de 9,5%, dont un peu moins de la moitié sont survenus entre la deuxième et la cinquième année.
Dans l’analyse, les prédicteurs d’un plus haut risque entre la deuxième et la cinquième année étaient la présence d’une cause athéroscléreuse de l’AVC- cette maladie qui bouche les artères du cœur et du cerveau par un dépôt de cholestérol- ou d’une cause embolique d’origine cardiaque (l’arythmie cardiaque étant la plus fréquente de ces causes), ou un score de risque élevé (score mixant la présence d’une hypertension artérielle, d’un diabète, d’un âge de plus de 60 ans, d’une durée de l’épisode initial supérieur à 10 minutes, ou la présence d’une paralysie ou d’un trouble du langage dans les symptômes de l’AIT).
Après un AIT ou un infarctus cérébral mineur ne laissant pas de handicap, le risque de refaire un AVC handicapant ou un infarctus du myocarde, fatals ou non, est de 6,4% la première année et de 6,4% entre la deuxième et la cinquième année. Ce résultat a été obtenu alors que tous les patients dans cette étude ont été traités de façon optimale, c’est-à-dire suivant les recommandations de traitement après un AVC.
Les auteurs préconisent de développer des stratégies de prévention encore plus efficaces pour diminuer le risque d’AVC. Parmi celles-ci, figurent de nouveaux médicaments comme ceux agissant sur le cholestérol, ou les triglycérides, ou encore des mesures d’hygiène simples comme l’exercice physique régulier (par exemple 20 à 30 minutes de vélo d’appartement tous les matins avant la douche) et la perte de poids. Comme seulement 25% des AVC sont précédés d’AIT, d’autres stratégies de détection des patients à risque devront être trouvées. La médecine connectée devrait pouvoir également contribuer à les identifier.
DOCUMENT inserm LIEN |
| |
|
| |
|
|
|
Une nouvelle canalopathie cérébrale associant déficience intellectuelle et mouvements anormaux |
|
|
| |
|
| |

Une nouvelle canalopathie cérébrale associant déficience intellectuelle et mouvements anormaux
COMMUNIQUÉ | 27 NOV. 2020 - 9H00 | PAR INSERM (SALLE DE PRESSE)
NEUROSCIENCES, SCIENCES COGNITIVES, NEUROLOGIE, PSYCHIATRIE
Les dysfonctionnements des canaux ioniques – ou canalopathies – dans le cerveau sont aujourd’hui associés à plus de 30 maladies neurologiques comme l’épilepsie ou encore les ataxies cérébelleuses. Structures situées sur la membrane des cellules permettant le passage d’ions (par exemple les ions sodium et potassium) entre l’intérieur d’une cellule et son environnement extérieur (milieu extracellulaire), ces canaux permettent notamment de générer et contrôler les potentiels d’action dans les neurones. Une étude menée à l’Institut du cerveau (Sorbonne Université/Inserm/AP-HP/CNRS) a permis d’identifier une nouvelle canalopathie cérébrale ayant pour origine des mutations dominantes du gène KCNN2, codant pour le canal ionique SK2. Les résultats ont été publiés dans Brain le 27 novembre 2020.
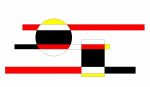
Les variants pathogéniques du gène KCNN2 identifiés chez les patients et leur localisation sur la structure protéique du canal SK2.
Les variant en rouge sont des variants pathogènes tronquant (introduisant un codon stop dans la séquence protéique). Les variants en noirs sont les variants pathogènes faux-sens associés à une perte de fonction. Le variant en gris a été classé de signification inconnue car le canal avec ce variant n’a pas montré de déficit particulier en électrophysiologie.
Le Dr Fanny Mochel, généticienne au sein du département de génétique de l’hôpital de la Pitié-Salpêtrière AP-HP et chercheuse à l’Institut du cerveau (Sorbonne Université/Inserm/AP-HP/CNRS) et le Pr Christel Depienne, généticienne à l’institut de génétique humaine de l’Hôpital Universitaire d’Essen (Allemagne) et également chercheuse à l’Institut du cerveau ont identifié un nouveau syndrome associé à des mutations du canal SK2. L’étude publiée dans la revue scientifique Brain porte sur 10 patients, 6 hommes et 4 femmes âgés de 2 à 60 ans présentant des retards intellectuels plus ou moins sévères associés, pour certains, à des troubles du spectre autistique ou des épisodes psychotiques. Ces troubles cognitifs sont dans tous les cas associés à des tremblements, à des symptômes d’ataxie cérébelleuse ou encore à des mouvements anormaux.
Grâce à une collaboration avec Agnès Rastetter de la plateforme de génotypage/séquençage de l’Institut du cerveau (Sorbonne Université/Inserm/AP-HP/CNRS), le génome d’un premier patient recruté à la Pitié-Salpêtrière a été analysé à la recherche de mutations génétiques à l’origine de ce syndrome. Cette analyse a mis en évidence une mutation du gène KCNN2 interrompant sa séquence codante, absente des parents du patient (mutation de novo). L’imagerie cérébrale par IRM (imagerie par résonance magnétique) chez ce patient a mis en évidence des anomalies de structure et d’intégrité de la substance blanche du cerveau, c’est-à-dire la gaine cérébrale protectrice des axones des neurones.
Par ailleurs, une collaboration internationale a permis aux chercheurs d’identifier 9 autres patients avec mutations du gène KCNN2. La majorité de ces mutations étaient survenues de novo tandis qu’une mutation était transmise dans une forme familiale du même syndrome.
Enfin, en travaillant conjointement avec Carine Dalle de la plateforme d’exploration cellulaire d’électrophysiologie de l’Institut du cerveau, les équipes des Dr Mochel et Depienne ont montré un rôle délétère de ces mutations sur la fonction du canal SK2, c’est-à-dire une perte de fonction entrainant un dysfonctionnement du canal ionique SK2 et donc une perte de régulation du potentiel d’action, support du message nerveux.
Les résultats de cette nouvelle étude ont permis d’identifier une nouvelle canalopathie cérébrale ayant pour origine des mutations dominantes du gène KCNN2, codant pour le canal ionique SK2. Ce nouveau syndrome se caractérise par la présence, d’une part, de symptômes cognitifs, en particulier une déficience intellectuelle et, d’autre part, de symptômes moteurs tels que des mouvements anormaux.
Cette nouvelle pathologie, dont on connaît maintenant la cause, est très hétérogène d’un point de vue des symptômes et nécessite une prise en charge multidisciplinaire à la frontière entre la génétique, pour la recherche des mutations du gène KCNN2, la neuropédiatrie et la neurologie pour la prise en charge des manifestations cognitives et motrices des patients.
DOCUMENT inserm LIEN |
| |
|
| |
|
|
|
Le rôle clé des astrocytes pour le développement cognitif |
|
|
| |
|
| |
Le rôle clé des astrocytes pour le développement cognitif
COMMUNIQUÉ | 01 JUIL. 2021 - 20H00 | PAR INSERM (SALLE DE PRESSE)
NEUROSCIENCES, SCIENCES COGNITIVES, NEUROLOGIE, PSYCHIATRIE
Les astrocytes sont des cellules du cerveau qui ont longtemps été considérées uniquement comme de simples cellules de soutien des neurones. Depuis quelques années, elles sont de plus en plus étudiées, et leur importance pour le fonctionnement du cerveau est peu à peu mis au jour. Des chercheurs de l’Inserm, du CNRS et du Collège de France au Centre interdisciplinaire de recherche en biologie révèlent désormais le rôle crucial de ces cellules dans la fermeture de la période de plasticité cérébrale qui suit la naissance. Les astrocytes auraient une place centrale dans le développement des facultés sensorielles et cognitives après la naissance. A plus long terme, ces travaux permettent d’envisager de nouvelles stratégies pour ré-introduire la plasticité cérébrale chez l’adulte, et ainsi favoriser la rééducation après des lésions cérébrales ou des troubles neuro-développementaux. Ces travaux ont été publiés dans la revue Science.
La plasticité cérébrale est une période transitoire clé où le cerveau, après la naissance, remodèle le câblage des neurones en fonction des stimulations extérieures qu’il reçoit (environnement, interactions…). La fin – ou fermeture – de cette période marque la stabilisation des circuits neuronaux, associée à un traitement efficace des informations et à un développement cognitif normal. Cela ne signifie pas qu’il n’y a plus aucune plasticité ensuite, mais qu’elle est très réduite par rapport au début de la vie.
Les problèmes qui interviennent pendant la période de plasticité cérébrale peuvent avoir des conséquences importantes à long terme. Ainsi par exemple, si durant cette période un individu souffre d’une pathologie oculaire qui l’empêche de voir correctement, comme par exemple un strabisme, le câblage cérébral qui correspond à cette faculté sera altéré définitivement si l’œil n’est pas soigné à temps.
Afin d’y remédier, les chercheurs ont pour objectif de remodeler ce câblage en identifiant une thérapie qui permettrait de réintroduire la plasticité cérébrale même après la fin du développement. Pour cela, ils cherchent aussi à mieux caractériser les mécanismes biologiques qui sous-tendent la fermeture de la période de plasticité cérébrale.
Des études pionnières des années 1980 ont montré que greffer des astrocytes immatures dans le cerveau d’animaux adultes permettait d’induire à nouveau une période de grande plasticité. L’équipe de la chercheuse Inserm Nathalie Rouach au Centre interdisciplinaire de recherche en biologie (Inserm/CNRS/Collège de France)[1] s’est inspirée de ce procédé pour révéler le processus cellulaire, jusqu’ici inconnu, à l’origine de la fermeture de la période de plasticité.
La greffe d’astrocytes immatures pour réintroduire la plasticité cérébrale
A travers des expériences menées en s’intéressant au cortex visuel de la souris, les chercheurs montrent que la présence des astrocytes immatures est clé pour la plasticité cérébrale. Les astrocytes orchestrent ensuite plus tard dans le développement la maturation d’interneurones[1] pendant la période de plasticité, ce qui aboutit in fine à sa fermeture. Ce processus de maturation a lieu via un mécanisme inédit impliquant l’action de la Connexine 30, une protéine que les chercheurs ont retrouvée en forte concentration dans les astrocytes matures durant la période de fermeture.
Le fait de greffer des astrocytes à des souris adultes pourrait-il permettre réintroduire une plasticité cérébrale ?
Afin de répondre à cette question, les chercheurs ont mis en culture des astrocytes immatures issues du cortex visuel de jeunes souris (qui avaient entre 1 et 3 jours). Ils ont ensuite greffé ces astrocytes immatures dans le cortex visuel primaire de souris adultes. Il s’agissait alors d’évaluer l’activité du cortex visuel après quatre jours d’occlusion monoculaire, une technique classique pour évaluer la plasticité cérébrale. Les chercheurs ont alors trouvé que la souris greffée avec des astrocytes immatures présentait un haut niveau de plasticité, contrairement à la souris non greffée.
« Cette étude nous rappelle qu’en neurosciences nous ne devons pas uniquement nous intéresser aux neurones. Les cellules gliales, dont les astrocytes font partie, régulent la plupart des fonctions du cerveau. Nous avons réalisé que ces cellules ont des rôles actifs. Les cellules gliales sont en effet moins fragiles que les neurones et constituent donc un moyen plus accessible d’intervenir sur le cerveau. », souligne Nathalie Rouach, coordinatrice de l’étude.
Les cellules gliales représentent plus de la moitié des cellules du cerveau. Elles n’ont pas le même lignage cellulaire que les neurones et leurs fonctions sont très différentes. On pensait jusque récemment qu’elles étaient les « nettoyeuses » du cerveau, mais les chercheurs ont réalisé qu’elles avaient aussi un rôle actif de libération de molécules. Par rapport aux neurones, elles arrivent plus tard dans le développement, n’ont pas le même mode de communication, et sont majoritaires.
Ces travaux sur les astrocytes permettent d’envisager de nouvelles stratégies cellulaires et moléculaires visant à ré-ouvrir une période de plasticité accrue chez l’adulte afin par exemple de favoriser la réadaptation après une lésion cérébrale ou de pallier les dysfonctionnements sensori-moteurs ou psychiatriques issus de troubles neuro-développementaux
[1] Les interneurones établissent des connexions entre un réseau de neurones afférent (qui envoie les informations au système nerveux central) et un réseau de neurones efférents (qui envoient ces informations vers les organes répondant à la stimulation)
DOCUMENT inserm LIEN |
| |
|
| |
|
|
|
Les cellules souches sanguines ont une mémoire immunitaire et ouvrent des pistes dans la recherche sur le Covid-19 |
|
|
| |
|
| |
Les cellules souches sanguines ont une mémoire immunitaire et ouvrent des pistes dans la recherche sur le Covid-19
COMMUNIQUÉ | 12 MAI 2020 - 9H15 | PAR INSERM (SALLE DE PRESSE)
BIOLOGIE CELLULAIRE, DÉVELOPPEMENT ET ÉVOLUTION | GÉNÉTIQUE, GÉNOMIQUE ET BIO-INFORMATIQUE | IMMUNOLOGIE, INFLAMMATION, INFECTIOLOGIE ET MICROBIOLOGIE
Les cellules souches du sang auraient une propriété surprenante. En plus d’assurer le renouvellement continu des cellules sanguines, ces cellules gardent une trace des infections passées pour déclencher une réponse immunitaire plus rapide et plus efficace par la suite, d’après une nouvelle étude co-dirigée par la chercheuse Inserm Sandrine Sarrazin et par le chercheur CNRS Michael Sieweke du Centre d’immunologie de Marseille-Luminy (CNRS/Inserm/Aix-Marseille Université) et du Centre des thérapies régénératives de l’Université technique de Dresde (Allemagne). Cette découverte pourrait avoir un impact significatif sur les futures stratégies de vaccination, notamment celles explorées dans le cadre de la pandémie de Covid-19. Elle permettrait aussi de faire progresser la recherche sur de nouveaux traitements visant à moduler le système immunitaire. Ces résultats ont été publiés dans la revue Cell Stem Cell.
C’est un fait connu de longue date : le système immunitaire adaptatif a une mémoire. Ainsi, les lymphocytes deviennent spécifiques d’un agent pathogène particulier à éliminer après y avoir été exposés lors d’une infection et certains d’entre eux subsistent durablement dans l’organisme. Les principes de la vaccination reposent sur la connaissance de ces mécanismes immunitaires.
Plus récemment, des travaux ont suggéré que le système immunitaire inné, qui permet la défense de l’organisme de façon immédiate suite à une infection, a lui aussi une forme de mémoire. Des chercheurs ont par exemple montré que le système immunitaire inné continue d’être plus efficace en cas de réinfection malgré la durée de vie très courte des cellules immunitaire, comme les monocytes ou les granulocytes. Ils ont alors soupçonné que cette mémoire du système immunitaire inné était en fait inscrite dans les cellules souches sanguines, dont la durée de vie est très longue, et qui sont à l’origine de différentes cellules immunitaires matures.
Pour vérifier cette hypothèse, les chercheurs du Centre d’immunologie de Marseille-Luminy (CNRS/Inserm/Aix-Marseille Université) et du Centre des thérapies régénératives de l’Université technique de Dresde (Allemagne) ont effectué des travaux dont les résultats sont publiés dans Cell Stem Cell. L’équipe a exposé des souris à une molécule de surface de la bactérie E. coli (lipopolysaccharide ou LPS), un agent pathogène largement utilisé pour mimer des infections en laboratoire.
Ensuite, les chercheurs ont transféré des cellules souches sanguines prélevées chez ces animaux à d’autres souris non infectées et dont le système immunitaire avait préalablement été détruit. Le but était de reconstituer entièrement leur système immunitaire à partir de ces cellules souches.
Les chercheurs ont ensuite infecté des souris de ce groupe avec une bactérie vivante de l’espèce P. aeruginosa et ont constaté que le taux de mortalité n’était que de 25 %. Il atteignait en revanche 75 % chez des souris contrôles, dont les cellules souches n’avaient jamais été exposées à un agent pathogène.
« Ce travail démontre de façon forte que les cellules souches sanguines ont une fonction de mémoire qu’on ne soupçonnait pas. Une première exposition à un pathogène les arme pour mieux affronter une prochaine infection», explique Sandrine Sarrazin.
Ce mécanisme n’est pas spécifique d’un agent pathogène puisque, dans une autre expérience, une première exposition des cellules souches sanguines à un antigène viral a protégé les souris contre une exposition secondaire à P. aeruginosa. De manière surprenante, les scientifiques ont donc découvert que la protection apportée par cette mémoire du système immunitaire s’étend au-delà du seul agent infectieux utilisé pour la première infection.
Les chercheurs se sont ensuite intéressés à la manière dont cette mémoire était codée. En étudiant le génome des cellules souches sanguines des souris infectées, ils ont constaté des modifications durables dans son organisation spatiale. Ces changements étaient susceptibles de modifier l’expression de certains gènes impliqués dans la réponse immunitaire innée. « Lors du premier contact avec l’agent pathogène, des gènes requis pour la réponse immunitaire sont en fait durablement mis en avant pour activer rapidement le système immunitaire lors d’une deuxième infection», explique Bérengère de Laval, première auteure de l’étude. Enfin, l’équipe a recherché des molécules impliquées dans ce changement de structure du génome et a découvert qu’une protéine appelée C/EBP bêta jouait un rôle majeur.
Des recherches pertinentes dans la lutte contre le Covid-19 ?
Ces résultats résonnent tout particulièrement en cette période de pandémie du coronavirus SARS-Cov-2.
Des observations récentes suggèrent que le vaccin BCG, connu pour induire lui aussi une mémoire immunitaire innée, agirait également au niveau des cellules souches sanguines et offrirait un certain degré de protection contre les infections respiratoires. Des études sont en cours pour tester son utilité contre le Covid-19.
Les découvertes de l’équipe pourraient éclairer les mécanismes en jeu dans cette protection au niveau moléculaire et ouvrir de nouvelles pistes vaccinales, y compris contre le Covid-19.
« Nos découvertes représentent une contribution majeure à la compréhension de la mémoire du système immunitaire et des fonctions des cellules souches du sang. Elles orientent en outre vers de nouvelles stratégies pour stimuler ou limiter la réponse immunitaire dans divers états pathologiques, et pourraient permettre d’affiner les stratégies de vaccination actuelles pour une meilleure protection face à divers agents pathogènes, y compris contre le SARS-CoV-2 », espère Michael Sieweke.
DOCUMENT inserm LIEN |
| |
|
| |
|
| Page : [ 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65 66 67 68 69 70 71 72 73 74 75 76 77 78 79 80 81 82 83 84 85 86 87 88 89 90 91 92 93 94 95 96 97 98 99 100 101 102 103 ] Précédente - Suivante |
|
|
|
|
|
|